

How We Get New Drugs in the U.S.: Drug Development and Preclinical Testing
In medical education, certain aspects of clinical practice in the U.S. are, in my view, not discussed enough. Experiencing the “real world” revealed several components of the medical system I was unaware of. But at least I know all the enzymes in the Krebs Cycle...
One example is the U.S. drug development process. During their preclinical years, medical students devote immense amounts of time and energy to memorizing pharmacology. Providers reflexively prescribe common drugs— statins come to mind— without a second thought. But how do these medications originate? How do they materialize into a small pill in a patient's hand from a concept in a scientist’s mind?
The drug development pipeline in the U.S. is lengthy, intricate, and expensive; on average, it takes 14 years from the initial idea to market approval and costs around two billion U.S. dollars. Additionally, extensive federal regulations must be understood and adhered to throughout the process. Understanding this process is crucial to understanding clinical drug development and clinical research. But, many medical professionals remain unaware.
Through the upcoming posts, I aim to provide a high-level overview of the drug development and approval process in the U.S. This series will not be an exhaustive examination of the entire system; instead, I will cover the fundamentals and include links to sources for more detailed information.
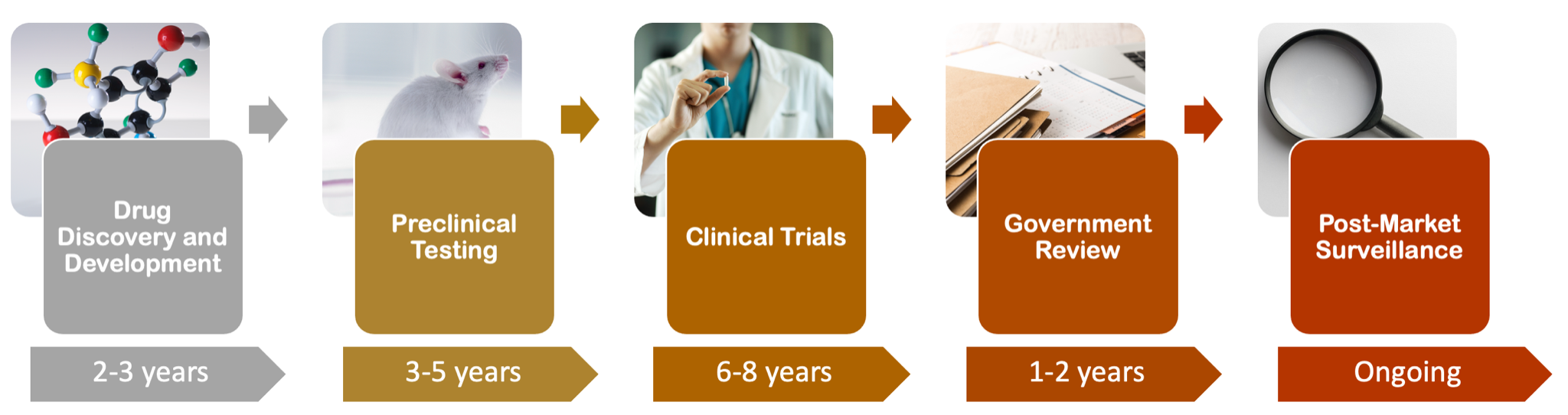
The drug development paradigm has five stages: drug discovery and development, preclinical testing, clinical trials, governmental review, and post-market monitoring. I have included Figure 1 here to clarify the overall procedure.
In future posts, I will cover additional aspects of this process, but here, I will concentrate on the first two stages: drug discovery/development and preclinical testing.
Drug Discovery and Preclinical Testing: Why?
These first two stages aim to find a drug candidate and collect enough safety and efficacy data to submit an Investigational New Drug (IND) application to the FDA. If the potential drug receives IND status, the entity that developed the drug—typically a pharmaceutical company—can proceed with human clinical trials to assess safety and efficacy. Therefore, producing strong data in the initial stages of drug development is essential for FDA approval and, ultimately, for the successful market introduction of a new drug. Let’s dive into these first two phases in more depth.
Drug Discovery and Development
How do we know which potential drugs to start with?
Historically, candidate drugs have been found by empirical observations of how compounds affect patient disease processes. This process of discovery—observing how a drug affects a patient and then determining how and why that drug works—is the reverse of modern drug discovery. The history of oncology drugs such as aminopterin demonstrates this paradigm.
In modern drug development, basic research first identifies altered molecular pathways in diseases, and compounds are then designed to modulate these pathways. Machine learning, high-throughput screening, and bioinformatics are utilized to efficiently find effective compounds with minimal side effects. For example, basic cancer research might uncover that malignant cells can alter how T cells interact with them, and this may represent a targetable process that a drug can modulate to allow T cells to kill cancer cells more effectively. This basic research is, therefore, the first step in drug development.
After basic research has identified a general area of interest, the next step is to identify a concrete molecular target—a process called target identification. Using the same T-cell example, researchers may identify PD-L1, a cell surface protein that can put the brakes on an immune response, as a target on which a potential drug may act.
After a target has been identified, an appropriate molecule is developed to interact with it. This molecule—called a “lead”—is expected to alter the disease process, and, not surprisingly, this step is known as lead identification. The lead can then be chemically altered—lead optimization—to create a compound that produces the maximum effect with the fewest undesired side effects. I’ve illustrated these steps in Figure 2.
Once the lead is discovered and optimized, the next step is determining its impact on a living system.
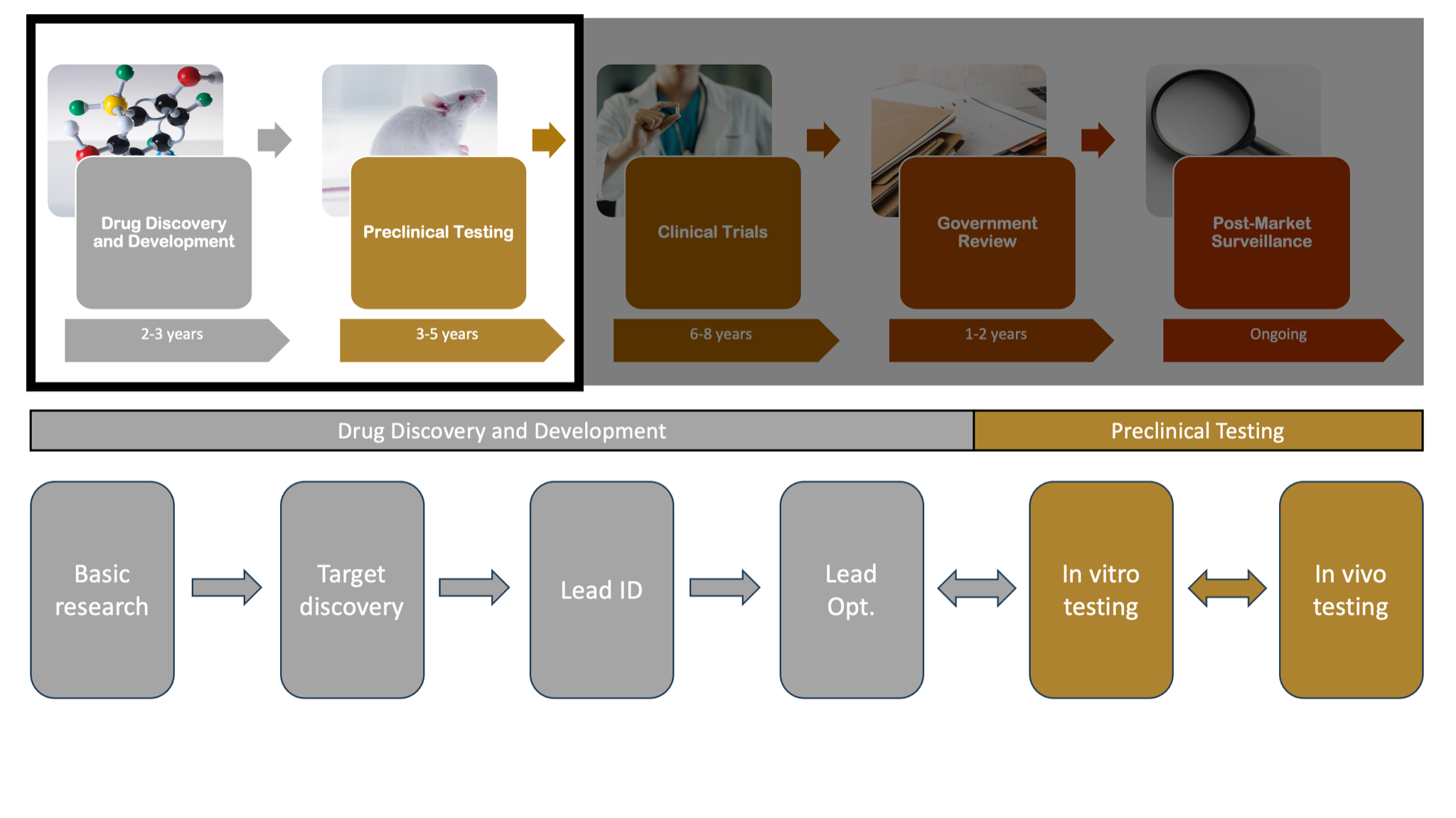
Preclinical Testing
Most people are familiar with this part of the preclinical process: The potential drug is injected into a cell culture dish or given to a mouse, and the reaction is measured. Do the cells survive? Does the mouse have a weird response at the injection site? Or is the tumor obliterated?
Although computer modeling can identify targets and design potential leads, it cannot, at this point, determine how that lead will affect the disease process or the side effects it will cause in a living system. Therefore, after the lead has been identified and optimized, it must be tested in living biological systems. This can be done using either in vitro or in vivo models or, more commonly, a combination of both. In-vitro models, typically conducted in a glass tube or culture dish, involve testing the lead using methods such as cell culture. This may include injecting the lead into a cell culture plate with cancer cells to see if and at what dose the cancer cells begin to die. In contrast to in-vitro models, in vivo models involve testing the lead in animals ranging from tiny mice to large primates.
Since 2022, the FDA has no longer required data from animal models in preclinical testing. Due to ethical considerations, many pharmaceutical and device companies invest in developing more robust computer software or complex in-vitro studies. For example, 3D cell culture models and other tissue engineering techniques have been of interest recently.
However, animal models provide researchers with the most adequate data on how a lead behaves in a living system, which correlates most strongly with how that lead will behave in human subjects. Therefore, many companies still use animals for preclinical testing.
Using these models, researchers hope to discover several critical features of the lead, such as:
- Effectiveness against the disease process
- Overall toxicity
- Side effects
- Pharmacokinetics (how the drug is absorbed and distributed in the body)
- Pharmacodynamics (biochemical and physiologic effects of the drug)
- Route of administration
- Reproductive toxicity
- Carcinogenicity
- Dosing strategy (amount and route)
The preclinical testing stage interacts closely with the lead optimization step. A lead with an undesired effect in a living system can be further optimized to reduce those effects and tested again.
Remember, all these steps—in both the drug design and preclinical testing stages—are designed to help a pharmaceutical company submit an IND application to the FDA so that their new drug can be tested in human clinical trials.
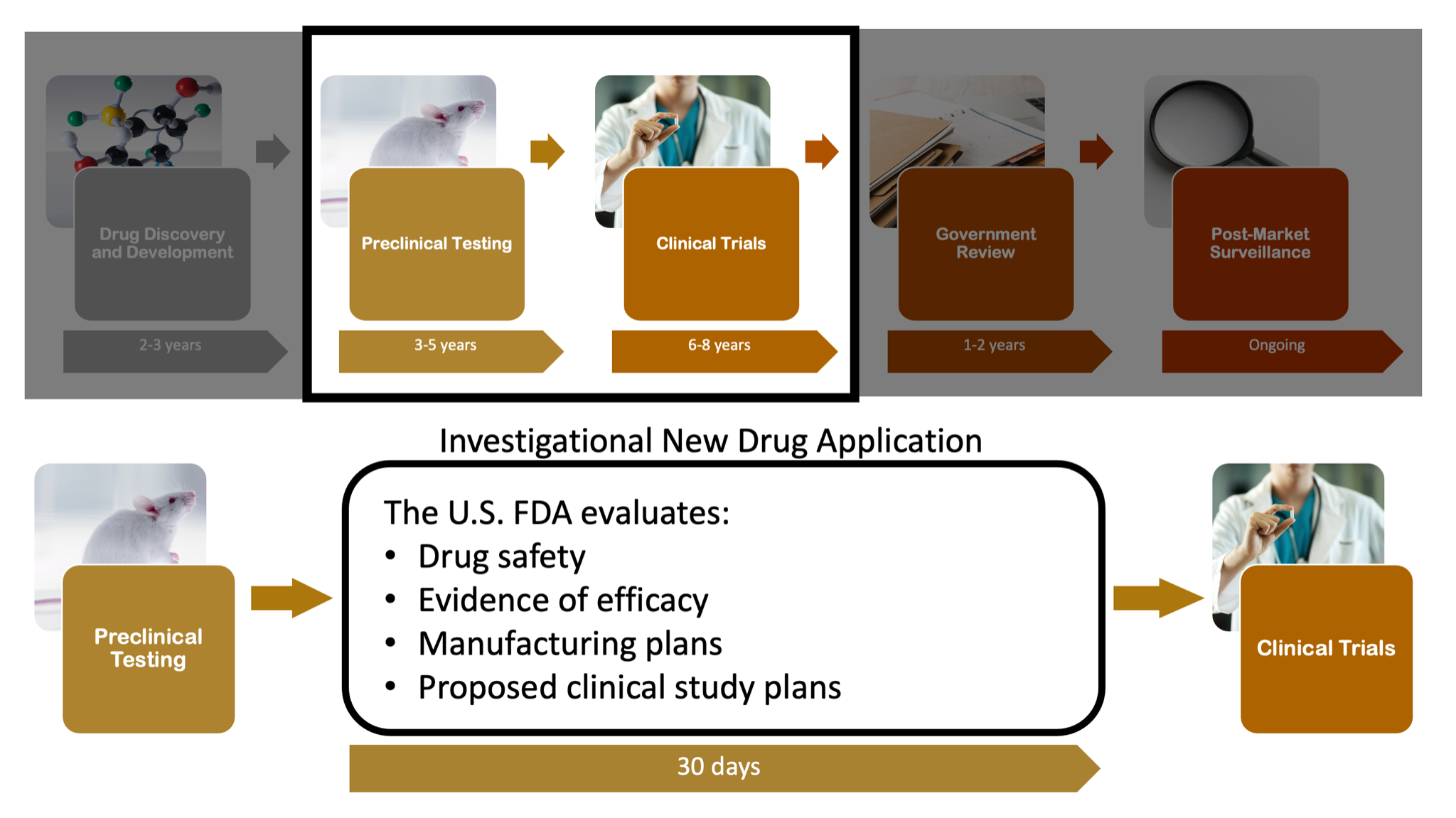
The Investigational New Drug Application
The IND application is a complicated multi-modular document that is submitted to the FDA, and includes preclinical study data meant to demonstrate a few components of the drug, such as:
- How effective the drug is
- How safe it is
- How is it going to be manufactured and stored
- How the proposed clinical trials are designed
- How the drug will be promoted and marketed
Here is a video that discusses the various components of the actual IND application in greater detail.
After submitting the IND application, the FDA typically reviews it for about 30 days to determine whether it is safe and effective for clinical trials. During this time, FDA reviewers may contact the sponsor with questions concerning specific aspects of the application.
After 30 days, there are two potential outcomes:
The FDA may impose a “clinical hold” on the IND application if concerns arise. This can occur if there are issues regarding safety, efficacy, the clinical trial protocol, or other areas of development. To lift the clinical hold, the sponsor must resolve these concerns that the FDA has.
But, if the FDA finds that the drug is safe and effective and a solid clinical trial plan is in place, the IND will be classified as “active.” This allows the sponsor to initiate testing on humans during clinical trials.
I’ve simplified this IND process in Figure 3.
This overview simplifies the first two stages and the IND application process. While the subject is more intricate, possessing a high-level understanding of this initial part is beneficial. I hope this post provides that. Also, the resources linked here will offer additional insight for those interested in learning more. The details provided here definitely far surpass what I knew about this process as a medical trainee, but at least I know the intricate details of the immune complement system...
Stay tuned for my upcoming post on clinical trials and how they are designed to evaluate the drug's safety and efficacy in humans.
For a Deeper Dive:
Here is an excellent review article on the drug development process and preclinical testing
The FDA has published a series of YouTube videos on the regulatory process involved in the drug discovery and preclinical testing phases.
Post a comment